Condrosarcoma
Estudio del condrosarcoma
Publicado en: Revista Española de Cirugia Osteoarticular
Tomo 22. Mayo-Junio de 1987
Valencia
RESUMEN
Se hace una revisión bibliográfica del condrosarcoma con motivo de la malignación de una osteocondomatosis múltiple en la pelvis.
Descriptores: Condosarcoma. Osteocondroma múltiple.
SUMMARY
A bibliographical review of the chondrosarcoma is reported. A case of malignant transformation os the pelvic multiple cartilaginous exotosis is described.
Key words: Chondrosarcoma. Pelvic multiple cartilaginous exostosis.
Introducción
Ante las molestias que presenta M.ª P.F. en forma de Dolores indefinidos y aumento súbito de tamaño del bultoma localizado en su zona glútea superior derecha, se nos plantea el establecer un diagnóstico y un pronostico.
Se trata de una paciente de 36 años, casada, con un hijo, que hasta la fecha había sido controlada por sus exótosis hereditarias multiples y que ahora ingresa para su estudio y tratamiento.
En la fase previa del estudio hay que destacar el antecedente de exótosis multiples hereditarias, la edad de la paciente- introducida en la cuarta década- y el aumento súbito y notorio del tamaño del bultoma, en un espacio de tiempo inferior a los seis meses y siendo de localización pélvica.
Con los datos radiológicos y con la sospecha, más que probable de malignidad, procedimos a su extirpación, llegándose al diagnóstico definitivo por anatomía patológica, de Condrosarcoma.
Condrosarcoma
Definición
Es un tumor maligno primitivo, que se origina a expensas del cartílago tumoral, y sus células forman cartílago pero no tejido óseo.
Hay que diferenciar el Condrosarcoma primario del Condrosarcoma secundario (11 por 100 de los casos según Dahlin) (2,3,5), que se originan a partir de un condroma u osteocondroma preexistente
Se distinguirá del condroma por la presencia de un tejido tumoral más celular y pleomórfico y por el número considerable de células voluminosas con núcleos grandes o dobles, según las definiciones de la O.M.S. (20,21)
Historia
En 1855 Wolkman inició el estudio descriptivo de los tumores cartilaginosos que prosiguió, en 1870, el profesor PAGET (17).
PHEMISTER, en 1930, (18) continuando el trabajo iniciado por Keiller, en 1925, descubrió la forma clínica que permitió diferenciar el Condrosarcoma del Osteosarcoma –éste es dos veces má frecuente que el primero-, no sólo se valió del aspecto clínicoradiológico y morfológico, sino que también valoró su curso más lento y su mejor pronóstico.
No obstante, THOMSON Y TURNER-WARWIICK (22), en 1955 sembraron un cierto confusionismo al basar su diferenciación en la predominancia del cartílago presente.
Fueron LICHTENSTEIN y JAFFE (15) quienes demostraron que el Condrosarcoma es una entidad totalmente distinta, fundamentando sus afirmaciones en el hecho de que <el Condrosarcoma se desarrolla a partir del cartílago completamente maduro>.
En 1959, LICHTENSTEIN y BRENSTEIN (14) describieron el Condrosarcoma mesenquimatoso, para ser Unni y Dahlin quienes definieron el Condrosarcoma de células claras en 1976.
Un año después, en 1977, SCHAJOWICZ (19) permitió, con sus trabajos, individualizar al Condrosarcoma yuxta-cortical.
Frecuencia
Los tumores cartilaginosos, ya sean benignos o malignos, son relativamente frecuentes. Representan un 25 por 100 según FEUILHADE DE CHAUVIN (10) y un 24 por 100 según DAHLIN (3,4).
Si nos limitamos a los tumores óseos malignos primitivos veremos como en unas estadísticas representan del 10-15 por 100 –S. Chavanne- y en otras del 12-25 por 100 –B. Tomeno- del total.
Es dos veces menos frecuente que el Sarcoma osteogénico y un poco más frecuente que los sarcomas de EWING (9) y de PARKER.
La relación entre las formas primitivas y las secundarias, así como la relación entre las formas centrales y periféricas varían ostensiblemente según quien sea el autor del estudio estadístico. LINCHTESTEIN y JAFFE (15) observaron un porcentaje idéntico entre las formas primitivas y secundarias. En cambio, Dahlin menciona en su estudio un 76 por 100 de formas primitivas y sólo un 24 por 100 de formas secundadias.
Según LINCHTESTEIN y JAFFE (15) la forma central sería de 2 a 5 veces más frecuente que la forma periférica, mientras que otros autores aseguran que la proporción entre la forma central y periférica es idéntica.
Sexo
Existe unanimidad en afirmar que la predominancia es masculina, aunque resulta difícil concretar el porcentaje.
Para unos, la predominancia masculina es 2/3 (23) y para otros, como Dahlin, es de algo menos del 60 por 100 del total.
Edad
El Condrosarcoma afecta a adultos y a personas de la tercera edad; traduciéndolo a números, encontramos que su predominancia oscila entre los 30-70 años, siendo la edad media de 45 años.
Según las estadísticas de DAHLIN (4), se ha encontrado un paciente afecto de Condrosarcoma que solamente contaba 3 años y 3 que tenían menos de 10 años.
Durante la segunda década de la vida, que es cuando hay más incidencia de Osteosarcoma, sólo se presenta un 3.8 por 100 del total de los Condrosarcomas.
Localización
Al contrario de los tumores cartilaginosos benignos que se desarrollan sobre todo en los extremos de los huesos pequeños, los condrosarcomas aparecen de manera preferente en la pelvis, escápula, esternón, costillas o extremos de húmero y fémur, es decir, en los huesos planos y zonas de gran riqueza en hueso esponjoso.
También se han hallado tumores condrosarcomatosos en el maxilar superior, que se supone proceden del cartílago de las paredes de las fosas nasales (1). Dahlin, en su estudio estadístico que concluyó en diciembre de 1975, encontró tres casos localizados en el hueso hioides y un caso de tumor primario de la cápsula y sinovial de la rodilla, sin signos de Condromatosis sinovial benigna previa (1).
Excepcionalmente se pueden hallar Condrosarcomas en cráneo y en los huesos pequeños de manos y pies (24).
En el Hospital Cochin (10) se han hallado Condrosarcomas en zonas poco frecuentes como son el antebrazo -3 casos- y en el peroné-2 casos-.
Asimismo, se puede mencionar que los Condrosarcomas hallados en el húmero y fémur suelen ser de tipo central, mientras que los localizados en pelvis son de tipo periférico (4).
Sintomatología
Como ya hemos apuntado, aparecen en adultos de mediana edad, llamando la atención un dolor indefinido y profundo, así como una importante tumefacción –son los síntomas iniciales más frecuentes-(3,4,6,23).
En algunas localizaciones, como la pelvis y el sacro, podemos hallar signos de compresión vásculo-nerviosa o visceral, que pueden presentarse de forma brusca al inicio de la enfermedad o durante el transcurso de la misma.
En las formas centrales el dolor precede a la aparición de la tumefacción que a su vez evoluciona lentamente, pero de manera progresiva. Por el contrario, en las formas periféricas es la tumefacción lo que atrae la atención del enfermo.
El tumor, durante un largo período de tiempo, puede ser indoloro, pero su volumen puede ir en aumento. Si el paciente tiene más de veinte años, iniciaremos la sospecha de malignidad ante un tumor cartilaginoso (6), delante de la persistencia de un dolor óseo profundo y, también, ante el aumento del volumen de la tumefacción.
Excepcionalmente, se pueden presentar fracturas patológicas en las formas centrales.
La evolución de la enfermedad variará según se trate de la forma periférica -10 años- o bien, de forma central-2 ó 3 años-, a excepción de casos muy malignos en los que la evolución puede ser de sólo un año y que representa del 5-10 por 100 del total de Condrosarcomas.
Radiología
El estudio radiográfico aportará unos datos que van a ocupar una posición de privilegio en el momento de realizar el diagnóstico ya que, generalmente, se observan imágenes patecnomónicas de Condrosarcoma(2,3,4,5).
Desde el punto de vista radiológico es aconsejable estudiar las formas centrales, periféricas y extraesqueléticas separadamente (23).
A.- Forma central.- El tumor se origina en la médula y permanece mucho tiempo limitado en esta zona.
La cortical se ve destruida tardíamente, el tumor invade, entonces, las partes blandas y tiende a englobar al hueso. No obstante, esta invasión podría producirse sin destrucción macroscópica de la cortical, por lo que no habría testimonio radiográfico evidente.
Cuando se ven afectados los huesos largos, el tumor, asienta en la metáfisis o en la diáfisis y, más raramente, en la epífisis (6).
1.- El primer tipo de Condrosarcoma central sería aquel que en las placas radiográficas se aprecia una geoda central, más o menos voluminosa uni o multilocular e irregular, con los bordes escleróricos bien definidos. La cortical es corroída progresivamente en forma de muesca.7algunos autores como Eideken y Hodes consideran la reacción ósea <endostal> como algo muy indicativo del diagnóstico de Condrosarcoma. Cuando la localización es en metáfisis o diáfisis de huesos largos, se produce un abombamiento fusiforme de la diáfisis, que se acompaña de engrosamiento de la cortical pero que, en este primer estadio es discreto.
Con bastante asiduidad, se aprecia en el interior de las geodas pequeñas masas calcificadas que recuerdan copos de nieve (6) y que “declaran” al Condrosarcoma y tiene un gran valor semiológico para defender la naturaleza cartilaginosa del tumor.
2.- El segundo tipo de Condrosarcoma central seria aquel que se hallaría en un estadio más avanzado que el anterior (6).
La corteza es destruida y el tumor invade las partes blandas.
No habrán bordes escleróticos bien definidos.
El volumen es variable y, en ocasiones, puede ser importante.
La reacción perióstatica se da con mucha frecuencia y es característico que contenga matriz condroide calcificada punteada.
B.- Forma periférica.- Tiene su origen, parece ser, en el casquete cartilaginoso de un Osteocondroma preexistente (4), sobre todo en los pacientes afectos de Osteocondromas hereditarios múltiples (1).- Fig 1-.
Estos tumores son muy insidiosos, lo que provoca que aumenten de tamaño, de forma notoria y rápida, siendo éste el motivo por el que al descubrirlos, el tamaño es ya importante. Sería este brusco crecimiento el que causaría el dolor.-Fig. 2-.
Mientras la cortical del hueso no se rompa a expensas de la exostosis, el dolor será sólo local.
Este tumor permanece siempre bien delimitado. Su aspecto es globuloso con sus contornos lobulados.- Fig. 3-.
Este tumor parece unido al hueso y presenta generalmente una condensación de la cortical en el punto de esta “unión”. Así mismo, en la masa tumoral, existen múltiples calcificaciones punteadas.
En el centro del tumor estas calcificaciones son más numerosas, más densas y más voluminosas, tienden a confundir. En la periferia se esfuman, se separan unas de las otras y son más difíciles de distinguir.-Fig.4-.
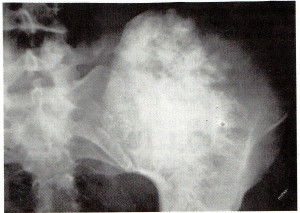
En un estadio más avanzado, a veces, el Condrosarcoma destruye o enmascara la exótosis a expensas de la que se origina, pero, también, en una etapa más avanzada el tumor tiene la capacidad destructiva. La cortical y la medular son invadidas por el tumor afectándose, al final, las partes blandes.
En algunos casos, los puntos de necrosis se ven calcificados de forma irregular, aunque algunos son en realidad verdadero tejido óseo.
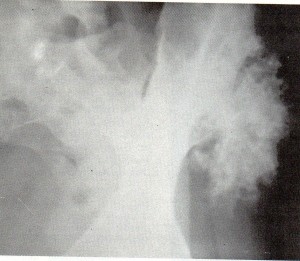
Figura 2
C.- Forma extraesquelética.- Según JAFFE (11) pueden formarse tumores de cartílago en tejidos blandos que no están en relación ni con hueso ni con cartílago normal. Aunque estos tumores suelen ser benignos, en algunos casos pueden dar lugar a Condrosarcomas.
Las imágenes radiológicas son inespecíficas si las lesiones carecen de calcificación. Sólo en el 30 por 100 de los casos hay calcificaciones cartilaginosas características y son más evidentes cuando existe erosión o infiltración del hueso.
Con cierta asiduidad los centros lobulares aparecen necrosados, licuados y quísticos.
En ocasiones, el Condrosarcoma tiene un aspecto mixoide, lo que nos permite asegurar un mal pronóstico (4).
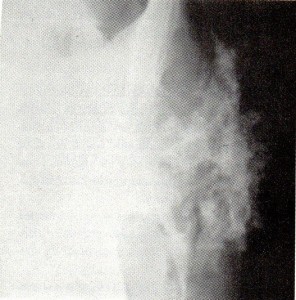
Figura 3
Histopatología
Es de todos conocida la dificultad diagnóstica para conseguir una concreta clasificación morfológica entre las lesiones cartilaginosas benignas y malignas. Así, a pesar de la evidente clínica existente en algunos Condrosarcomas, su etiquetamiento hispatológico es realmente difícil.
El estudio histopatológico, básicamente, nos debe permitir aclarar la verdadera identidad del Condrosarcoma y su nivel de malignidad.
LICHTENSTEIN y JAFFE (15) nos aconsejan, para establecer el diagnóstico diferencial basarnos en:
- La presencia de muchas células de núcleos gruesos
- La existencia de células con núcleos gruesos dobles
- La visión de células condrales gigantes con núcleo voluminoso o con masas de cromatina
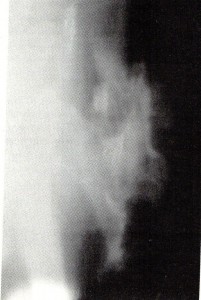
Figura 4
Por contra, hay autores que, siguiendo los trabajos de DAHLIN, O’NEAL y ACKERMAN (1&) y EVANS (8) dividen en tres grupos los distintos caracteres celulares que nos permiten establecer un diagnóstico histopatológico. Así nos hallamos delante de:
- Condrosarcoma muy diferenciado: Sus células, en ocasiones, multinucleadas o con un núcleo que ocupa 1/3 o más del citoplasma –lo que resulta difícil es detectar estas células tan significativas-.
- Condrosarcoma diferenciado: Su profileración celular está formada por condrocitos diferenciados, con núcleos unas veces densos y otras pálidos- en estas células no suelen producirse mitosis-.
- Condrosarcoma indiferenciado: Formado por células cartilaginosas indiferenciadas y globulosas, polinucleadas.
A pesar de estas amplias consideraciones, debemos ser prudentes en el momento de recoger las muestras, realizar los cortes y valorar el posible tejido neoplástico.
Otros exámenes diagnósticos
-La arteriografía: sus aportaciones diagnósticas son pocas, pondrña en evidencia una hipervascularización periférica no específica.
-La escintigrafía: aporta pocos datos
-La xerografía: permite visualizar los contornos de la parte no calcificada de un Condrosarcoma
-El scanner: permitirá una importante mejora en el estudio de la invasión del hueso y de las partes blandas.
Diagnóstico referencial
Desde el punto de vista radiológico:
- El Condrosarcoma central de asiento habitual en la metáfisis y/o diáfisis puede confundirse con:
- Infarto óseo, por las calcificaciones que hay en el centro de la lesión.
- Ciertas formas de displasia fibrosa
- Más raramente, con una lesión metastásica
- Un condroma benigno que presente signos de malignidad como son: una rotura de la cortical e invasión de partes blandas.
- El Condrosarcoma epifisario debe diferenciarse:
- Un tumor de células gigantes.
- Un condroblastoma.
- El condrosarcoma periférico, debe distinguirse de los otros tumores que aparecen sobre la cortical:
- De la exótosis osteogénica, relativamente fácil
- Del sarcoma yuxta-cortical (19), que aparece antes de los 30 años y su imagen es más densa, más homogénea y más limitada.
- Del sarcoma benigno periférico, será preciso recurrir a la Histología
Evolución
A.- Problemas locales
Lo más destacable es la aparición de recidivas locales. Suelen aparecer a los dos o tres meses de efectuado el tratamiento.
Algunos tumores con gran necrosis conllevan un alto riesgo de siembra peroperatoria, dando recidivas posteriores.
Según EVANS (8), el 92 por 100 de recidivas aparecen después de una escisión limitada; 15.6 por 100 aparecen después de amputación o resección amplia.
La topografía, también influye en el momento de aparecer las recidivas. Así el 46 por 100 se da en tumores que afectan al tronco, el 38 por 100 se da en los que afectan a las extremidades.
Por el contrario, parece que el grado de afectación histológica influye poco sobre el porcentaje de recidivas.
B. La metástasis
Son bastantes raras en este tipo de tumores, afectan al pulmón y suelen aparecer después de varios años de evolución.
Aquí su afección sí está en estrecha con relación con el grado histológico.
Según EVANS (8) en el grado 1 no hay metástasis, en el grado 2 sólo un 10 por 100 y en el grado 3 hay un 70 por 100.
Pronóstico
Se ve influenciado por:
- El lugar de asiento del tumor, así los tumores que asientan en el tronco son más peligrosos que los que asientan en las extremidades. La supervivencia a los 5 años es el 50 por 100 en los que asientan en el tronco y de un 70 por 100 en los que asientan en las extremidades.
- El tratamiento inicial. DAHLIN (5) apunta que el 69 por 100 de los enfermos tratados inicialmente con erradicación completa tienen una supervivencia a los 10 años, mientras que los demás ven reducido el porcentaje a un 18.6 por 100.
- El grado de su histología
Tratamiento
A.- Radioterapia: Es ineficaz en los tumores cartilaginosos. Sólo se usaría en caso extremo, como medida paliativa.
B.- Quimioterapia: No tiene indicación.
C.- Azufre radioactivo: Se administra por vía general y no parece ser capaz de conseguir la curación, aunque, en casos desesperados, puede usarse por su acción analgésica.
D.- La cirugía: Es prácticamente la única posibilidad terapéutica, pero sólo nos valdremos de la amputación o de la resección amplia.
La elección dependerá de las condiciones locales, el asiento, del tamaño del tumor, de la proximidad del paquete vásculo-nervioso y del grado de expansión del tumor.
Después de una resección se dan casos en los que la función del miembro es buena sin requerir otro acto quirúrgico reparador. En otros casos, será preciso estableces la función por artrodesis o por prótesis. Es en la pelvis donde se suelen hallar las mayores dificultades reparadoras (7).
Cuando la resección no da seguridad, es necesario recurrir a la amputación.
BIBLIOGRAFIA
1.- BROCHERIOU, C. et PEYEY, J (1975): Tumeurs cartilagineuses des maxillaires. Ann. Anat. Pathol. 20, n.º1, 23-34.
2.- DAHLIN, D.C. (1975):Bonne Tumors. Charles C. Thomas. Springfield, IV.
3.- DAHLIN, D.C. (1975):Bonne Tumors. Charles C. Thomas. Springfield (Ilinois)
4.-DAHLIN, D.C. (1980): Tumores óseos. 2ª Ed., 17, 180-204.
5.- DAHLIN, D.C and HERDERSON, E. D. (1962): Mesenchymal chondrosarcoma; further observations on a new entity cancer, 15:410.
6.- EIDEKEN,J.; HODES, P.J. (1978): Diagnóstico radiológico de las enfermedades de los huesos. 2º Ed. 15:963-974.
7.- ENNEKING, W.F. et DUNHAN, W.K. (1978): Resection and reconstruction for primary neoplasms, involving the innominate bone. J. Bone Joint Surg., 60-A, nº6,731-746
8.-EVANS, M.L.; AYALA, A.G.; ROHMSDAHL, M.D. (1977): Pronostic factors in chondrosarcoma of bone. A Clinicopatologic analysis with emphasis on histologic granding. Cáncer 40: 808-837.
9.- EWING, J.A. (1939): A review of the classification of bone tumors. Bull. Am. Coll. Surg., 24:290-295.
10.-FENILHADE DE CHAUVIN, P. (1978): Chondrosarcoma des membres. Thèses, Faculté de Medicine de Paris.
11.-JAFFE, H.L. (1945): Hereditary multiple exótosis. Arch Pathol., 36:355
12.- JAFFE, J. L. (1958): Tumors ans tumorous conditions of the bones and joints. Lea&Feliger, Philadelphia.
13.- KEILLER, V.A. (1925): Cartilaginous tumors of bone. Surg. Gynecol. Obstret., 40: 510-521.
14.-LINCHTENSTEIN, L.; BERMSTEIN, D. (1959): Unusual benign and malignant chondroid tumors of bone. Cancer 12:1142-1157.
15.-LICHTENSTEIN, L. and JAFFE, M. L. (1943): Chondrosarcoma of bone. An J. Pathol., 19:533-589.
16.-O’NEAL, L.W. y ACKERMAN, L.V. (1952): Chondrosarcoma of bone. Cancer, 5:551-577.
17.-PAGET, J. (1870): Lectures on surgical Pathology, ed. 3 p. 489. Longmans, Green and Co. London.
18.-PHEMISTER, D.B. (1930): Chondrosarcoma of bone. Surg. Ginecol. Obst., 50:216.
19.- SCHAJOWICZ, F. (1978) Juxta cortical chondrosarcoma. J. Bone Joint Surg. (Br), 59:473-480.
20.- SCHAJOWICZ, F. (1982): Tumores y lesions seudotumorals de huesos y articulaciones. 169-214.
21.-SCHAJOWICZ, F. and BESSONE, J.F., (1967): Chondroscroma in 3 brothers a pathological and genetic study. J. Bone Joint Surg., 49-A:129.
22.-THOMSON, A. D.; TURNER-WARWICK, R. T. (1955): Skeletal sarcomata and giant-cell tumor. J. Bone Joint Surg. (Br) 37:266-303
23.-TOMESCO, B.; FENILHADE DE CHAUVIN, P. et HONCKE, M. (1980): Chondrosarcome. Encyclop. Méd. Chir. Paris. App. Loc., 140303 D-20, 11.
24.-TRIAS, A.; BASORA, J. ; SÁNCHEZ, G. y MADARMAS, P. (1978): Chondrosarcoma of the hand. Clin. Orthop., 134, 297-301.
25.-UNNI, K. K.; DAHLIN, D. C.; BEABOUT, J. W. (1976): Periosteal osteogenic sarcoma. Cancer, 37: 2476-2485.